Vaskulitiden sind eine Gruppe von Erkrankungen, die durch Vaskulitis, Ischämie und Schäden an den von den betroffenen Gefäßen versorgten Organen gekennzeichnet sind. Die betroffenen Arterien Arterien Arterien sind unterschiedlich groß und lokalisiert und variieren je nach Art der Vaskulitis. Vaskulitiden können eine primäre Erkrankung oder sekundär zu einer anderen Grunderkrankung sein. Es gibt keine eindeutig bekannte Pathophysiologie. Die Diagnose sollte bei jeder Person mit tastbarer Purpura, Lungeninfiltraten, ischämischen Ereignissen und Multisystemerkrankung in Betracht gezogen werden. Eine rechtzeitige Erkennung und Therapie der Vaskulitiden ist zwingend erforderlich, da es sich oft um schwerwiegende und manchmal tödliche Erkrankungen handelt. Die Behandlung umfasst immunsuppressive, antivirale und/oder entzündungshemmende Mittel.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Vaskulitiden sind Erkrankungen der Blutgefäße mit Entzündungen der Gefäßwände, die zu Blutungen, Ischämie und Nekrose des distalen Gewebes und der Organe führen.
Die folgende Klassifikation wurde aus der Nomenklatur der primären Vaskulitiden der International Chapel Hill Consensus Conference 2012 übernommen:

Purpura:
A: Diffuse kleine tastbare Purpura an den unteren Extremitäten
B: Vergrößerte Ansicht
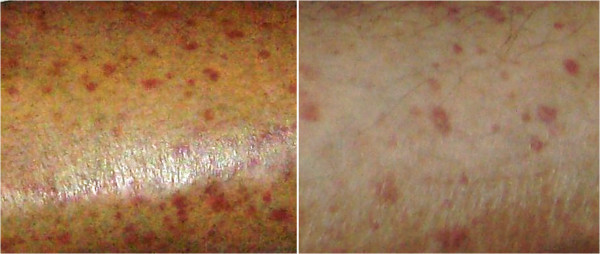
Purpurischer Hautausschlag bei einer Person mit kryoglobulinämischer Vaskulitis als Folge einer Bindegewebserkrankung.
Bild : „Purpuric skin rash“ von Gheita TA et al. Lizenz: CC BY 4.0
Mehrere Ulzerationen bei einer Person mit mikroskopischer Polyangiitis
Bild : „Skin ulcers revealing microscopic polyangiitis“ von Khammassi N et al. Lizenz: CC BY 2.0
Angiographie der Takayasu-Arteriitis:
Beachten Sie die Stenose und poststenotische Dilatationen in den Hauptästen des Aortenbogens.
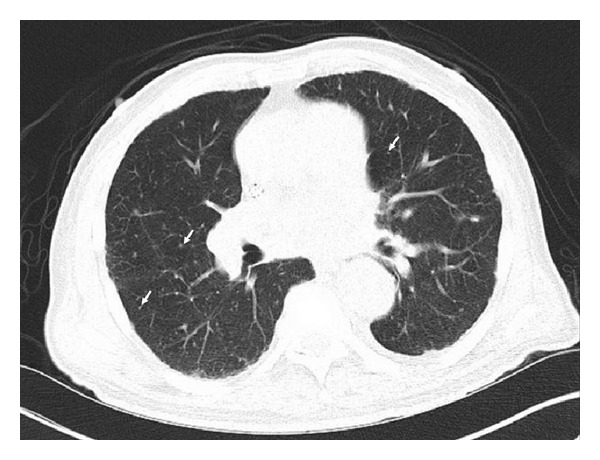
CT-Scan mit diffusen kleinen bilateralen Lungenknötchen (Pfeile) als Folge einer granulomatösen Erkrankung, insbesondere bei großen verkalkten Granulomen mit mediastinaler und hilärer Lymphadenopathie.
Die Person ist positiv für ANCA und diagnostiziert mit mikroskopischer Polyangiitis.
Die Hautbiopsie zeigt bei einer Person mit Polyarteriitis nodosa eine perivaskuläre lymphozytäre und neutrophile Infiltration mit fibrinoiden Nekrose der Gefäßwand und leukozytoklastischen und roten Blutkörperchen-Extravasationen.
Bild : „Skin biopsy shwoing perivascular lymphocytic and neutrophilic infiltration“ von Rodrigo D et al. Lizenz: CC BY 2.0



















