Hypokoagulopathien, auch bekannt als Blutungsstörungen oder Blutungsdiathesen, sind eine vielfältige Gruppe von Krankheiten, die zu einer abnormalen Hämostase führen. Hämostase ist der angeborene, schrittweise Prozess, der zur Blutstillung aus einem beschädigten Blutgefäß führt. Die physiologische Hämostase hängt von der Integrität der Endothelzellen und der subendothelialen Matrix, der Thrombozyten Thrombozyten Thrombozyten und der Gerinnungsfaktoren ab. Hypokoagulopathien resultieren aus Anomalien bei einem oder mehreren dieser Akteure, was zu unwirksamen thrombotischen Plaques und somit Blutungen führt. Die häufigste Blutungsstörung ist die Von-Willebrand-Krankheit.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Hypokoagulopathien, auch bekannt als Blutungsstörungen oder Blutungsdiathesen, sind eine vielfältige Gruppe von Krankheiten, die zu einer abnormalen Hämostase und einem erhöhten Blutungsrisiko führen.
Die physiologische Koagulation ist abhängig von der normalen Struktur und Funktion von:
Im Folgenden eine Zusammenfassung der Hämostase:
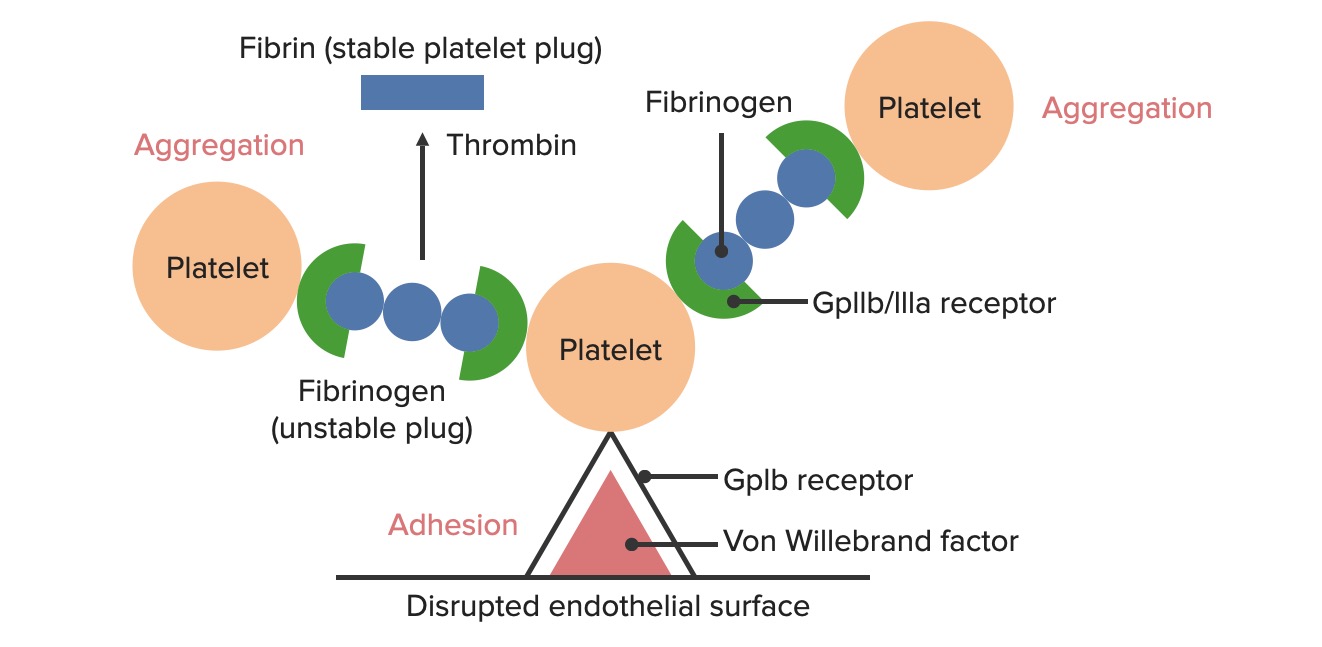
Bildung der temporären hämostatischen Aggregation:
Die zerstörte endotheliale Oberfläche setzt den von-Willebrand-Faktor (vWF) dem passierenden Blut aus. Thrombozyten binden über ihre GpIb-Rezeptoren an den vWF und werden aktiviert. Die Thrombozytenaktivierung veranlasst sie zur Sekretion von Adenosindiphosphat (ADP), das die Expression der GpIIb/IIIa-Rezeptoren auf den Thrombozyten stimuliert. Die GpIIb/IIIa-Rezeptoren binden an Fibrinogen, das an jedem Ende einen Thrombozyten binden kann, wodurch die Thrombozyten aggregieren. Wenn mehr Thrombozyten aneinander gebunden werden, wird ein Thrombozytenaggregat erzeugt. Wenn die Gerinnungskaskade aktiviert wird, wandelt Thrombin das schwächere Fibrinogen in das stärkere Fibrin um, wodurch ein viel stabileres Gerinnsel entsteht.

Übersicht der Gerinnungskaskade
a: aktiviertes Formular
PF3 : Thrombozytenfaktor 3 (Phospholipide)

Überblick über die Gerinnungskaskade
Bild von Lecturio.Die folgenden Bedingungen können zu einem hypokoagulierbaren Zustand führen.
| Kongenitale Bedingungen | Erworbene Bedingungen | |
|---|---|---|
| Gefäßwanderkrankungen |
|
|
| Thrombozytenerkrankungen |
|
|
| Gerinnungsstörungen | Hämophilie Hämophilie Hämophilie A, B und C |
|
| Gemischte Erkrankungen | Von-Willebrand-Krankheit | Disseminierte intravasale Koagulopathie |
| Medikamente |
|
|
Eine Hypokoagulopathie kann sich wie folgt darstellen:
| Hautblutungen | Schleimhautblutungen | Innere Blutungen | Blutungsbeginn nach Trauma | |
|---|---|---|---|---|
| Thrombozytenerkrankungen | ✓ | ✓ | – | Früh (sofort) |
| Gefäßerkrankungen | ✓ | – | – | – |
| Störungen der Gerinnungskaskade | – | – | ✓ | Verspätet (Minuten bis Stunden) |
Blutungsstörungen können aufgrund von Anomalien der Gefäßwand auftreten.
Angeborene Störungen der Bindegewebsmatrix können Gefäßbrüchigkeit verursachen, die zu häufigen Gefäßverletzungen führt.

Hereditäre hämorrhagische Teleangiektasien
Bild: „Clinical manifestations of HHT” von Latino et al. Lizenz: CC BY 4.0 , bearbeitet von Lecturio.Die folgenden erworbenen Erkrankungen können zu Gefäßbrüchigkeit führen und Gefäßverletzungen verstärken:
Hereditäre Bedingungen:
Erworbene Bedingungen:




















