Die Akromegalie und der Großwuchs werden durch eine übermäßige Produktion von Wachstumshormon (Englisches Akronym: GH) durch die Hypophyse Hypophyse Hypophyse verursacht. Hierbei kommt es zusätzlich zu einem Überschuss des Insulin-like growth factor 1 (IGF-1), der ebenso für die Klinik entscheidend ist. Als Hauptursache für die beiden Erkrankungsbilder gelten somatotrope Hypophysenadenome. Weiterhin möglich sind, wenn auch selten, extrahypophysäre Erkrankungen. Erkranken Betroffene bereits im Kindesalter, so kommt es aufgrund noch nicht verschlossener Wachstumsfugen zum Großwuchs, der durch eine besonders große Körperlänge auffällt, die 2 m überschreiten kann. Bei einem Erkrankungsbeginn im Erwachsenenalter, der deutlich häufiger ist, kommt es hingegen zur Akromegalie. Hierbei sind nur bestimmte Körperstrukturen vergrößert, da die Wachstumsfugen bereits verschlossen sind. Diagnostisch wegweisend sind neben der typischen Klinik die Labordiagnostik und die Bildgebung. Therapiemöglichkeiten sind chirurgisch, medikamentös oder strahlentherapeutisch.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Die Regulation erfolgt stufenweise :

Schematische Darstellung der direkten und indirekten Wirkungen des Wachstumshormons (GH)
Bild von Lecturio. Lizenz: CC BY-NC-SA 4.0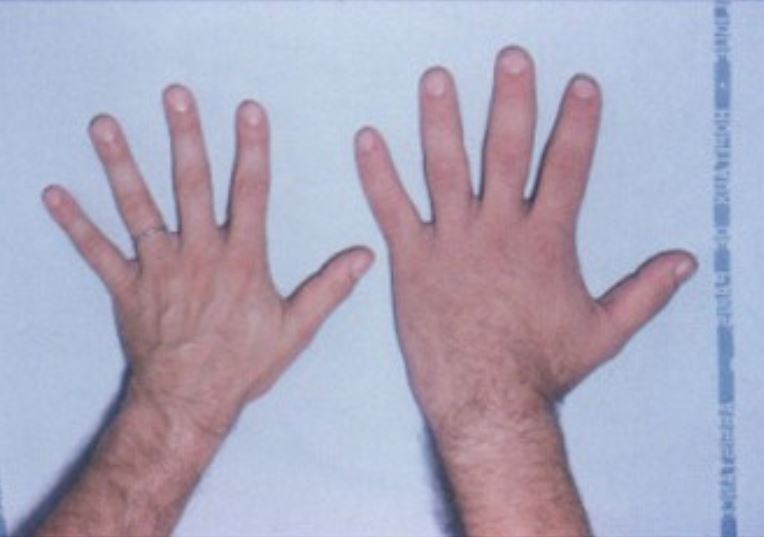
Die Hand eines Gesunden (links) neben der Hand eines Betroffenen mit Akromegalie (rechts)
Bild : “Acromegaly hands” von Philippe Chanson und Sylvie Salenave. Lizenz: CC BY 2.0
Darstellung eines Patienten mit den pathognomonischen Gesichtszügen einer Akromegalie: vergrößerte Nase, supraorbitale Wülste, Prognathie und ein großer Kopf
Bild : “Acromegaly facial features” von Philippe Chanson und Sylvie Salenave. Lizenz: CC BY 2.0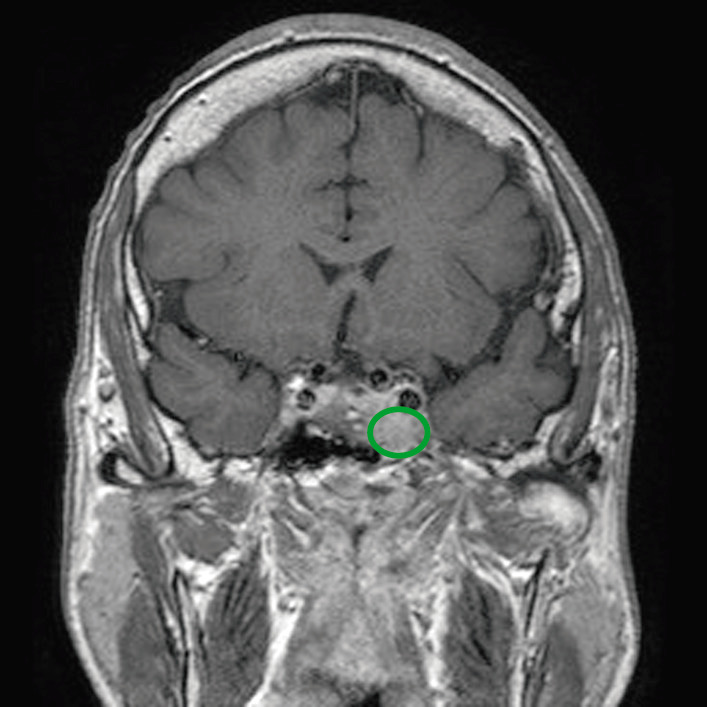
MRT einer betroffenen Person mit Hypophysenadenom (grüne Umrandung), das raumfordernde Effekte auf die umgebenden Strukturen hat
Bild : “Before pasireotide therapy. MRI scan of pituitary – coronal view” von Rajesh Rajendran et al. Lizenz: CC BY 3.0, bearbeitet von Lecturio.
Magnetresonanztomografie (MRT) einer betroffenen Person mit großen Hypophysenadenom, das zur Akromegalie führt
Bild : Acromegaly” von Elgee. Lizenz: CC BY 3.0

















