Das Marfan-Syndrom ist eine genetische Erkrankung mit autosomal-dominanter Vererbung. Es beeinflusst die Elastizität des Bindegewebes im ganzen Körper, insbesondere im Herz-Kreislauf-, Augen- und Bewegungsapparat. Auch Haut Haut Haut: Aufbau und Funktion, Lunge Lunge Lunge: Anatomie und das zentrale Nervensystem Nervensystem Nervensystem: Aufbau, Funktion und Erkrankungen sind betroffen. Die Patient*innen sind normalerweise groß und haben lange Gliedmaßen, Finger und Zehen sowie hypermobile Gelenke. Assoziierte Zustände sind Aortenaneurysma oder -dissektion, Mitralklappenprolaps Mitralklappenprolaps Mitralklappenprolaps und Linsenluxation. Die Diagnose wird klinisch nach festgelegten Kriterien gestellt, und Gentests werden nur dann durchgeführt, wenn sie das Management beeinflussen können. Die medizinische oder chirurgische Behandlung basiert auf klinischen Manifestationen. Eine kardiovaskuläre Beteiligung wird engmaschig verfolgt, da sie die Haupttodesursache ist.
Kostenloser
Download
Lernleitfaden
Medizin ➜

Familienmitglieder mit Marfan-Syndrom
Bild : „Family members with Marfan’s syndrome” von Birjand Atherosclerosis and Coronary Artery Research Centre, Birjand University of Medical Sciences, Birjand, Iran. Lizenz: CC BY 3.0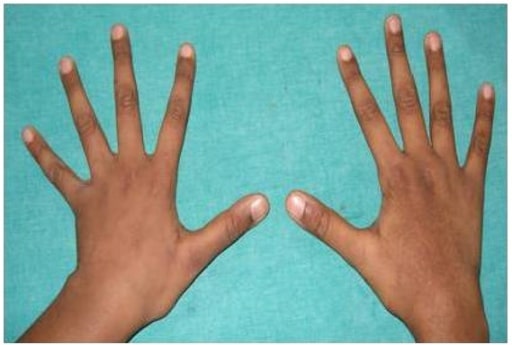
Foto zeigt Arachnodaktylie
Bild : „Arachnodactyly” von Department of Pedodontics, SRM Dental College, SRM University, Chennai . Lizenz: CC BY 3.0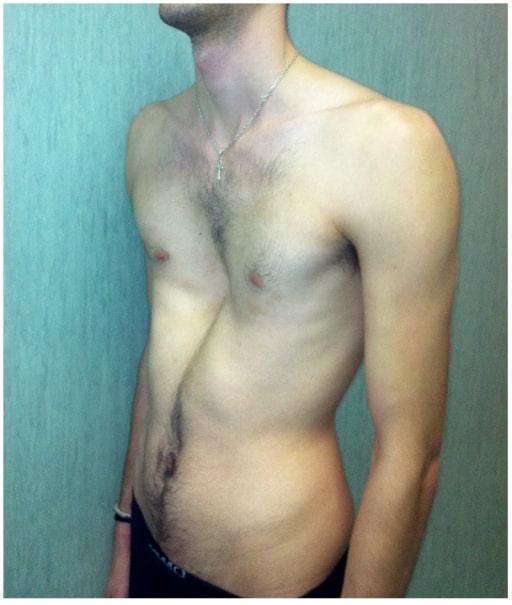
Schwerer Pectus Excavum bei einem vom Marfan-Syndrom betroffenen Mann
Bild : „fig2“ von Fernando De Maio et al. Lizenz: CC BY 4.0
Ein vom Marfan-Syndrom betroffener Mann mit Skoliose zusätzlich zu dorsalen Hautstreifen und reduzierter Ellbogenstreckung
Bild : „fig5“ von Fernando De Maio et al. Lizenz: CC BY 4.0
Ein Patient mit Marfan-Syndrom mit einem damit verbundenen Handgelenk- und Daumenzeichen:
(a) Das „Handgelenkszeichen“ ist positiv, wenn der Daumen beim Greifen des kontralateralen Handgelenks den fünften Finger überlappt.
(b) Das „Daumenzeichen“ ist positiv, wenn der Daumen weit über den ulnaren Rand der Hand hinausragt, wenn er von den Fingern überlappt wird.

Eine Armspanne größer als die Körpergröße des Patienten und ein hochgewölbter Gaumen
Bild : „Arm span more than height and high arched palate” by Consultant, Eye and Glaucoma Care, Gariahat, West Bengal, India. Lizenz: CC BY 3.0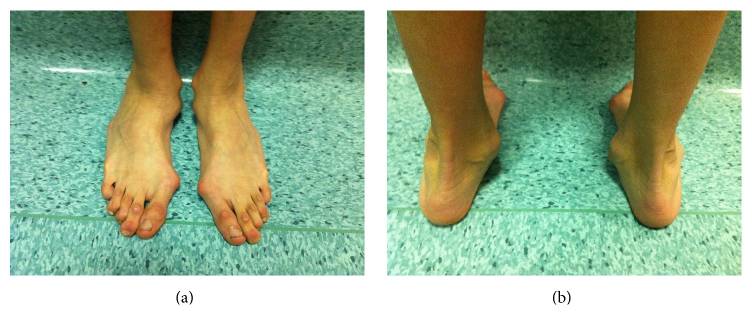
Rückfußdeformität mit ausgeprägter Valgusferse bei einem vom Marfan-Syndrom betroffenen Mann
Bild : „Hindfoot deformity“ von Fernando De Maio et al. Lizenz: CC BY 4.0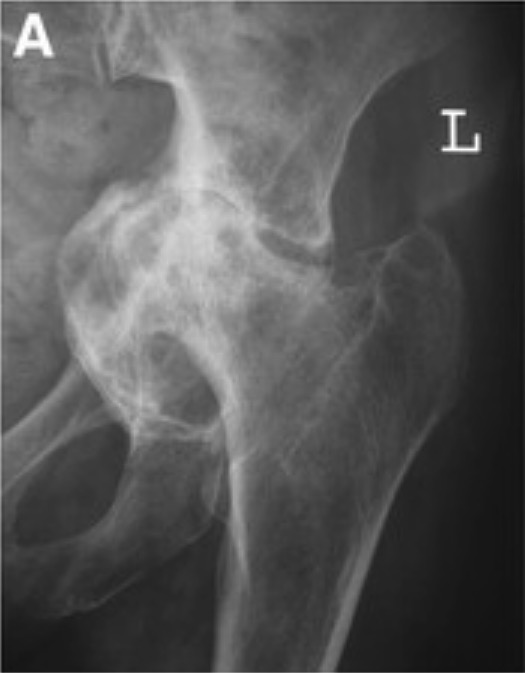
Röntgenbild mit Protrusio acetabuli: Man beachte, dass der Hüftkopf und die Pfanne in das Becken hineinragen.
Bild : „Grade III protrusio acetabuli“ von Orthopaedic Research Fellow, Royal Infirmary of Edinburgh, Little France EH16 4SA, UK. Lizenz: CC BY 2.0 , herausgegeben von Lecturio.
Makropathologisches Präparat eines Aortendissektionslappens und der Aortenwand
Bild : „Aortic dissection flap and aortic wall” von Department of Obstetrics and Gynaecology, Women’s and Children’s Hospital, Adelaide, Australia. Lizenz: CC BY 3.0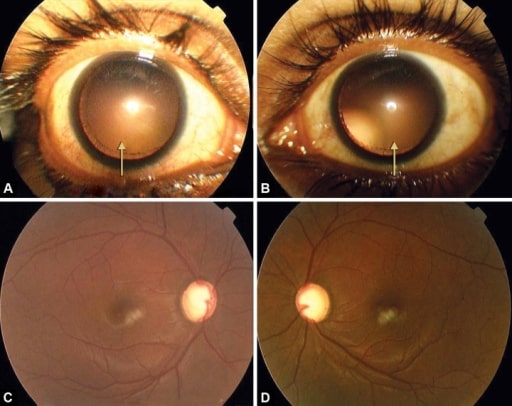
A und B: symmetrische temporale und nach oben gerichtete Subluxation der Linse bei Dilatation
C und D: fortgeschrittenes glaukomatöses Schröpfen auf beiden Augen
Bestandteile der Diagnosestellung:
Gent-Kriterien
Für die Diagnose des Marfan Syndroms:
oder
oder
Das Nichterfüllen der Kriterien schließt ein MFS allerdings nicht aus.

Transthorakale Echokardiographie mit Darstellung eines Intimarisses in der dilatierten Aortenwurzel oberhalb des Aortenklappenniveaus:
A: In der Systole ist ein linearer Intimariss direkt über der Aortenklappe zu sehen.
B: In der erweiterten Aortenwurzel ist ein linearer Intimalappen sichtbar.
Die Behandlung des MFS richtet sich nach den klinischen Manifestationen und sollte individuell bestimmt werden. Ein multidisziplinäres Team aus Kardiologen, Augenärzten, Orthopäden und Chirurgen wird benötigt.

Präoperatives Röntgenbild eines Teenagers mit Marfan-Syndrom und Skoliose
Bild : „Figure 1“ von Levy et al. Lizenz: CC BY 4.0
Postoperatives Röntgenbild desselben Patienten
Bild : „Figure 4“ von Levy et al. Lizenz: CC BY 4.0
Kardiales MRT der Aorta, das die Standard-Bildgebungslandmarken für die thorakoabdominale Aorta zur Überwachung zeigt: Dieses Bild zeigt die Dilatation der mittleren Aorta ascendens (3).
Bild : „F11“ des Manchester Heart Centre, Manchester Royal Infirmary, Oxford Road, Manchester M13 9WL, UK. Lizenz: CC BY 2.0



















