Die Chronische lymphatische Leukämie (CLL) ist eine hämatologische Neoplasie, die durch eine übermäßige Produktion von monoklonalen B-Lymphozyten im peripheren Blut gekennzeichnet ist. Sie wird nach der WHO-Klassifikation den niedrig-malignen-Non-Hodgkin-Lymphomen zugeordnet. Die Krankheit tritt normalerweise bei älteren Patient*innen mit einem Durchschnittsalter von 70 Jahren auf. Klinisch zeigt sich die CLL häufig asymptomatisch. Die Erkrankung kann daher nach zufälligem Befund einer Lymphozytose Lymphozytose Lymphozytose nach einer Blutentnahme und weiterer Diagnostik diagnostiziert werden. Bei Vorliegen von Symptomen sind die typischen Symptome: Lymphadenopathie Lymphadenopathie Lymphadenopathie, B-Symptomatik und Infektanfälligkeit. Im Blutausstrich fallen viele kleine Lymphozyten Lymphozyten Lymphozyten auf, die häufig Grumprecht-Kernschatten aufweisen. Eingeteilt wird die CLL in 3 Stadien nach Binet, die eine Aussage über die Prognose der Erkrankung treffen. Diese Einteilung basiert auf dem Ausmaß der Lymphadenopahie, dem Hämoglobinwert und der Thrombozytenzahl. Die Behandlung kann von „watchful waiting“ in frühen, asymptomatischen Stadien bis hin zu einer Chemotherapie reichen.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Die Chronische lymphatische Leukämie (CLL) gehört zu den niedrig-malignen B-Zell-Lymphomen und ist gezeichnet durch die Akkumulation funktionell beeinträchtigter Lymphozyten Lymphozyten Lymphozyten, oft monoklonalen Ursprungs im Blut.
Die CLL und das kleinen lymphatische Lymphom (SLL) wurden durch die WHO zu einer Krankheit zusammengefasst, die beide als Erscheinungsformen von B-Zell-Neoplasien gelten:
Die Hämatopoese Hämatopoese Knochenmark: Zusammensetzung und Hämatopoese beginnt mit einer hämatopoetischen Stammzelle, die durch entsprechende chemische Reize ( hämatopoetische Wachstumsfaktoren Hämatopoetische Wachstumsfaktoren Hämatopoetische Wachstumsfaktoren) zur Teilung und Differenzierung veranlasst wird.

Hämatopoese des Knochenmarks – Proliferation und Differenzierung der gebildeten Blutelemente:
Aus einer hämatopoetischen Stammzelle kann ein lymphoider oder myeloider Vorläufer entstehen. Die lymphatische Zelle kann zu einem oder mehreren Lymphozyten heranreifen: T-Zellen, B-Zellen oder natürliche Killerzellen (NK-Zellen). Die chronische lymphatische Leukämie tritt auf, wenn es zu Mutationen in Lymphozyten, insbesondere B-Zellen, kommt. Dadurch reifen Lymphozyten nur teilweise aus, sodass ihre Wirksamkeit beeinträchtigt ist.
KBE-GEMM: koloniebildende Einheit – Granulozyten, Erythrozyten, Monozyten, Megakaryozyten
CFU-GM: koloniebildende Einheit – Granulozytenmakrophagen
GM-CSF: Granulozyten-Makrophagen-Kolonie-stimulierender Faktor
M-CSF: Makrophagen-Kolonie-stimulierender Faktor
G-CSF: Granulozyten-Kolonie-stimulierender Faktor
NK: Natürliche Killerzellen
TPO: Thrombopoietin EPO: Erythropoetin
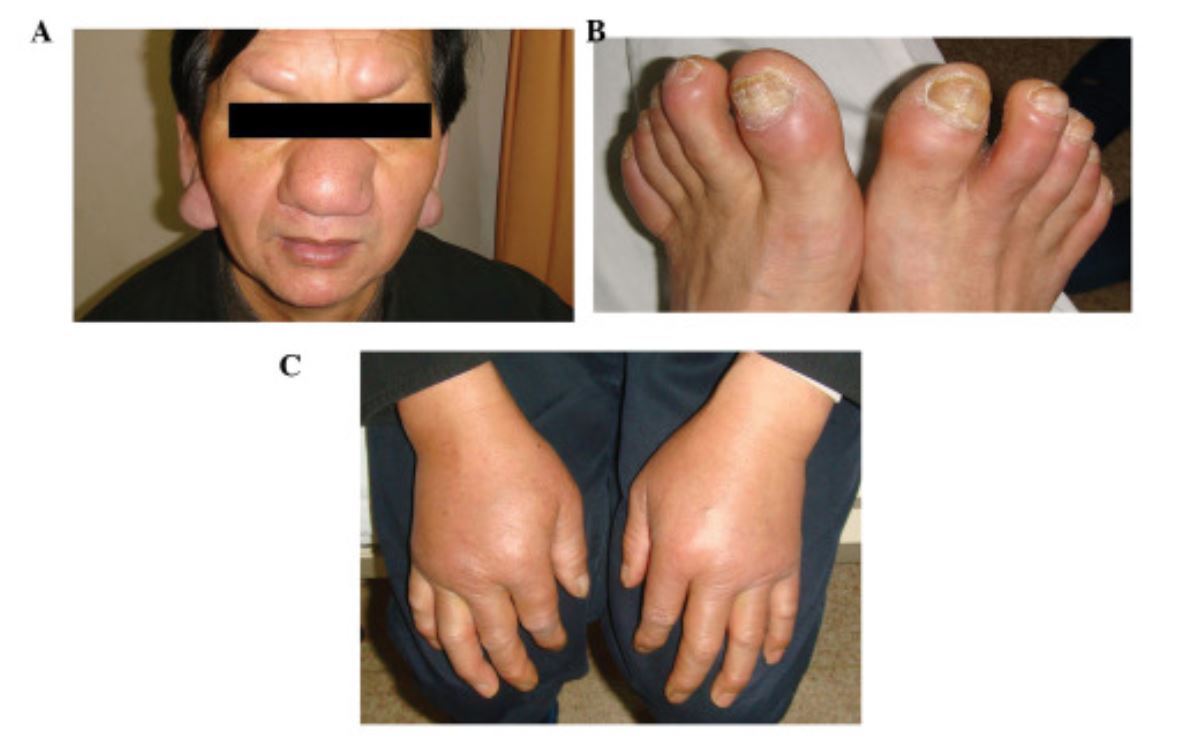
Seltene CLL-Infiltrationen der Ohren, Augenbrauen und Nase (A) und der Zehen (B). Bilaterales, nicht narbiges Ödem an den Handrücken (C).
Bild: „Cutaneous manifestations in a patient with chronic lymphocytic leukemia involving the head, neck and distal extremities“ von Lu C, Li L, Qiao Q, Liu G, Fang L. Lizenz: CC BY 3.0
Blutausstrich eines erwachsenen Mannes mit ausgeprägter Lymphozytose:
Die vorherrschenden Lymphozyten haben ein sehr spärliches, blasses Zytoplasma, runde bis leicht ovale Kerne und keine erkennbaren Nukleolen. Außerdem sind Gumprecht’sche Kernschatten zuerkennen, die für die Mehrheit der CLL-Fälle charakteristisch sind.
Die Stadieneinteilung nach Binet basiert auf der Anzahl betroffener Lymphknoten-Regionen und dem Vorliegen einer Anämie Anämie Anämie: Überblick und Formen/Thrombopenie und teilt drei Gruppen mit unterschiedlicher Prognosen ein. Zu den Lymphknoten-Regionen gehören (Lymphknotenvergrößerung 1 cm):
| Stadium | Beschreibung | Prognose |
|---|---|---|
| A (geringes Risiko) | ≤ 2 Regionen vergrößert | >10 Jahre |
| B (mittleres Risiko) | ≥ 3 Regionen vergrößert | 5-7 Jahre |
| C (hohes Risiko) | Vorliegen einer Anämie Anämie Anämie: Überblick und Formen (Hb <10 g/dl) oder Thrombozytopenie Thrombozytopenie Thrombozytopenie ( Thrombozyten Thrombozyten Thrombozyten <100.000/µl) | ca. 3 Jahre |
Dieses Klassifizierungssystem wird in den USA verwendet und wird in den deutschen Leitlinien nur erwähnt. Die Stadieneinteilung nach Rai erfolgt anhand des Vorliegens einer Lymphadenopathie Lymphadenopathie Lymphadenopathie, Hepato-/ Splenomegalie Splenomegalie Splenomegalie und einer Anämie Anämie Anämie: Überblick und Formen und Thrombozytopenie Thrombozytopenie Thrombozytopenie. Der Grenzwert für eine Anämie Anämie Anämie: Überblick und Formen ist bei diesem System strenger (<11 g/dl) als bei der Stadieneinteilung nach Binet.
| Risiko | Stadium | Beschreibung | Prognose |
|---|---|---|---|
| Niedrig | 0 | Lymphozytose Lymphozytose Lymphozytose im Blut oder Knochenmark Knochenmark Knochenmark: Zusammensetzung und Hämatopoese | > 150 Monate |
| Intermediär | ich | Lymphozytose Lymphozytose Lymphozytose + vergrößerte Lymphknoten Lymphknoten Lymphsystem | 101 Monate |
| II | Lymphozytose Lymphozytose Lymphozytose + vergrößerte Leber Leber Leber oder Milz Milz Milz mit oder ohne Lymphadenopathie Lymphadenopathie Lymphadenopathie | 71 Monate | |
| Hoch | III | Lymphozytose Lymphozytose Lymphozytose + Anämie Anämie Anämie: Überblick und Formen (Hämoglobin <11 g/dl) mit oder ohne Vergrößerung von Leber Leber Leber, Milz Milz Milz oder Lymphknoten Lymphknoten Lymphsystem | 19 Monate |
| IV | Lymphozytose Lymphozytose Lymphozytose + Thrombozytopenie Thrombozytopenie Thrombozytopenie (Thrombozytenzahl < 100.000/µl) mit oder ohne Anämie Anämie Anämie: Überblick und Formen oder vergrößerte Leber Leber Leber, Milz Milz Milz oder Lymphknoten Lymphknoten Lymphsystem |




















