Der Begriff „Störungen der sexuellen Entwicklung“ bezieht sich auf eine Gruppe von Erkrankungen, die durch eine atypische sexuelle Entwicklung eines Individuums gekennzeichnet sind, die Anomalien in der Struktur und/oder Funktion der inneren und/oder der äußeren Geschlechtsorgane beinhalten kann. Die typische Geschlechtsentwicklung beginnt mit dem chromosomalen Geschlecht (z. B. 46,XY oder 46,XX), das die sexuelle Differenzierung der Keimdrüsen Keimdrüsen Endokrines System: Überblick (z. B. Hoden Hoden Hoden oder Eierstöcke) bestimmt, die Hormone Hormone Endokrines System: Überblick ausschütten, die den Phänotyp (z. B. männlich oder weiblich) bestimmen. Die meisten Störungen der sexuellen Entwicklung sind auf Anomalien in bestimmten Chromosomen, Enzymen oder Rezeptoren Rezeptoren Rezeptoren zurückzuführen. Die Diagnose umfasst in der Regel die Analyse des Karyotyps und bestimmter Hormonspiegel. Die Behandlung kann komplex sein und umfasst häufig Psychotherapie Psychotherapie Psychotherapie, Hormonersatztherapie und/oder chirurgische Eingriffe.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Störungen der sexuellen Entwicklung sind eine Gruppe von Erkrankungen, die durch eine pathologische sexuelle Entwicklung eines Individuums gekennzeichnet sind und Anomalien in der Struktur und/oder Funktion sowohl der inneren als auch der äußeren Geschlechtsorgane aufweisen.
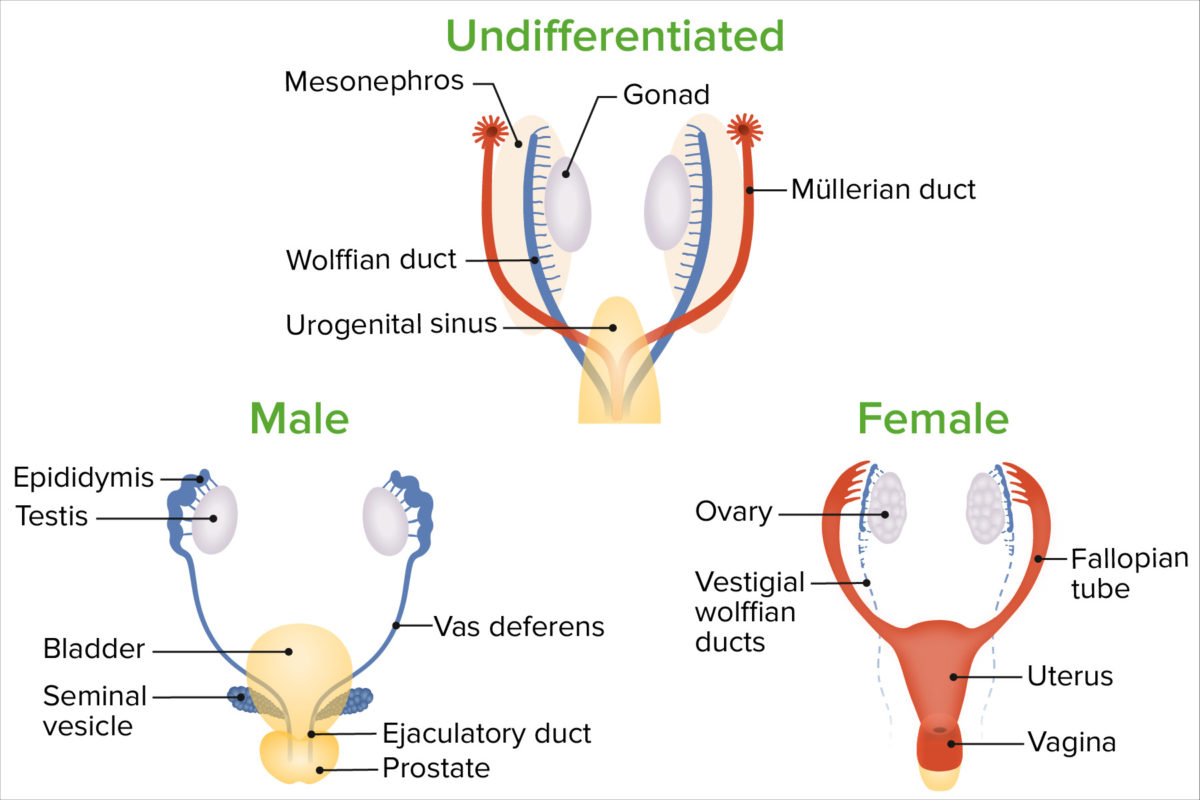
Geschlechtsdifferenzierung
Bild von Lecturio.
Phänotypische Differenzierung der äußeren Genitalien bei männlichen und weiblichen Embryonen
Bild von Lecturio.Die männliche Entwicklung beginnt aufgrund des Vorhandenseins des SRY-Gens.

Geschlechtsbestimmung beim Menschen: Das Y-Chromosom enthält das SRY-Gen (Sex-determining Region Y), das für den testis-determining factor (TDF) kodiert, der bewirkt, dass sich aus den embryonalen Keimdrüsen Hoden (männliche Keimdrüsen) bilden und eine männliche Entwicklung stattfindet. Fehlt das TDF-Protein (d. h. kein Y-Chromosom), entwickeln sich die embryonalen Keimdrüsen zu Ovarien (weibliche Keimdrüsen). Frauen besitzen zwei Kopien des X-Chromosoms (XX), Männer besitzen ein X- und ein (kürzeres) Y-Chromosom (XY).
Bild von Lecturio
Männliche Geschlechtsentwicklung
DHT: Dihydrotestosteron
| Störung | Pathophysiologie | Wichtige Punkte |
|---|---|---|
| Androgenresistenz Androgenresistenz Androgenresistenz (AIS) |
|
|
| 5-α-Reduktase-Mangel |
|
|
| Aromatase-Mangel Aromatase-Mangel Aromatase-Mangel |
|
|
| Adrenogenitales Syndrom Adrenogenitales Syndrom Adrenogenitales Syndrom |
|
|
| Kallmann-Syndrom Kallmann-Syndrom Kallmann-Syndrom |
|
|
| Gonadendysgenesie, 46, XX-Typ |
|
|
| Swyer-Syndrom Swyer-Syndrom Swyer-Syndrom |
|
|
| Echter Hermaphroditismus Hermaphroditismus Ovotestikuläre Störung der Geschlechtsentwicklung (Echter Hermaphroditismus) |
|
|
| Turner-Syndrom Turner-Syndrom Turner-Syndrom |
|
|
| Klinefelter-Syndrom |
|
|

21-Hydroxylase-Mangel:
Schematische Darstellung der Pathophysiologie des adrenogenitalen Syndroms aufgrund eines 21-Hydroxylase-Mangels
Die Differenzialdiagnose für Störungen der sexuellen Entwicklung umfasst in der Regel viele der oben genannten Erkrankungen. Die allgemeine diagnostische Vorgehensweise zur Bestimmung der richtigen Diagnose ist im Folgenden aufgeführt.

Androgenresistenz:
Eine 30-jährige Patientin mit primärer Amenorrhö. Bei der Untersuchung werden gut entwickelte Brüste und das Fehlen von Schamhaaren festgestellt, was auf ein Vorhandensein von Östrogenen ohne signifikante Testosteronspiegel hinweist.

Charakteristische Merkmale einer Frau mit Turner-Syndrom
Bild von Lecturio.Becken-Sonografie:
Erwägen einer Bildgebung der Nieren Nieren Niere und der Harnwege, da Anomalien der Geschlechtsorgane häufig mit Anomalien der Nieren Nieren Niere und der Harnwege verbunden sind.


















