Das Retinoblastom ist der häufigste Augentumor bei Kindern, aber insgesamt eine seltene Tumorerkrankung. Dieser maligne Netzhautumor geht von den retinalen Stammzellen aus. In den meisten Fällen wird das Retinoblastom durch eine genetische Mutation im Retinoblastom-Gen verursacht. Es kann aber auch zu einem sporadischen oder familiärem Retinoblastom kommen. Das Retinoblastom kann einseitig oder beidseitig vorkommen. Die Erkrankung macht sich typischerweise bei einem Kind unter 2 Jahren bemerkbar, als einseitige oder beidseitige Leukokorie (pathologische weiße Reflexion im Auge). Das Retinoblastom verläuft tödlich, wenn es nicht behandelt wird, aber bei früher Diagnosestellung und Therapieeinleitung liegt die Überlebensrate bei 95 %. Die Diagnose wird anhand ophthalmologischer Untersuchungen gestellt und mit einer apparativen Diagnostik ergänzt. Die Therapie ist Stadium-abhängig und umfasst verschiedene Behandlungsoptionen, wie Chemotherapie, Operation, Strahlentherapie, Kryotherapie oder Lasertherapie. Nach erfolgreicher Therapie sollte eine lebenslange Nachsorge erfolgen, da die Patient*innen ein erhöhtes Risiko für Zweitmalignome haben.
Kostenloser
Download
Lernleitfaden
Medizin ➜

Schema der beiden Formen der Retinoblastom-Entwicklung: links ein hereditäres Retinoblastom und rechts ein sporadisches Retinoblastom. Bei der erblichen Form wird ein mutiertes RB1-Gen (in blau) an das Kind weitergegeben, und es ist nur eine weitere Mutation im gepaarten normalen Allel (in rot) erforderlich, um den Tumor zu entwickeln. Bei der sporadischen Form kann das RB1-Gen in der Keimbahn der Nachkommen (in dieser Abbildung nicht dargestellt) de novo mutiert sein, sodass es sich dann wie in der vererbbaren Form verhält, oder eine retinale Vorläuferzelle kann eine RB1-Mutation in beiden Allelen erhalten und dadurch ein Retinoblastom entwickeln.
Bild von Lecturio.
Kind mit Retinoblastom des rechten Auges mit Leukokorie
Bild: “Pathology: Patient: Retinoblastoma” von The National Cancer Institute. Lizenz: Public Domain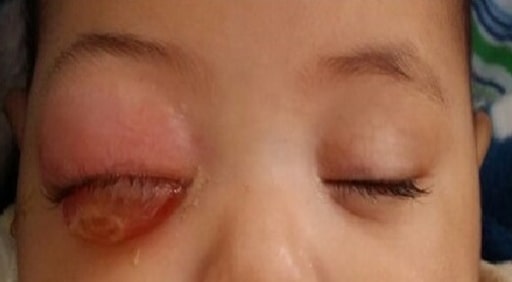
Kind mit orbitaler Zellulitis des rechten Auges aufgrund eines lokal fortgeschrittenen Retinoblastoms
Bild : “Orbital cellulitis” von The Pan African Medical Journal. Lizenz: CC BY 2.0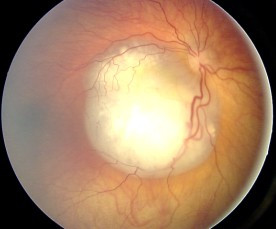
Augenhintergrund eines Retinoblastoms
Bild: “Fundus retinoblastoma” von Aerts, I, Lumbroso-Le Rouic. Lizenz: CC BY 2.0
Enukleiertes Auge mit großem exophytischen Retinoblastom
Bild: “Retinoblastoma” von The Armed Forces Institute of Pathology (AFIP). Lizenz: Public Domain
Retinoblastom mit charakteristischen Flexner-Wintersteiner-Rosetten mit einer kreisförmigen Anordnung kurzer säulenförmiger Zellen um ein zentrales Lumen
Bild: “Flexner-Wintersteiner rosettes” von The Armed Forces Institute of Pathology (AFIP). Lizenz: Public DomainDie Behandlung hängt vom Stadium ab, wobei mehrere Therapien zum Erhalten der Sehkraft verfügbar sind. Ziel ist das Überleben des Kindes und hoffentlich den Erhalt der Sehkraft.
Die Therapie von Retinoblastomen erfolgt meist in spezialisierten Kliniken. Wesentlich für die Prognose vor der Therapie ist die Ausbreitung des Tumors.
Es kann entweder Augapfel-erhaltend therapiert werden oder das Auge wird enukleiert.
Die Differentialdiagnosen umfassen alle Erkrankungen, die eine Leukokorie verursachen können.




















