Die Rapid progressive Glomerulonephritis Glomerulonephritis Membranoproliferative Glomerulonephritis (RPGN) beschreibt eine schwere glomeruläre Erkrankung mit fortschreitendem Verlust der Nierenfunktion innerhalb von Wochen bis Monaten. Die RPGN ist mit einem nephrotischen Syndrom verbunden und kann eine Manifestation verschiedener Krankheiten sein. Histologisch charakteristisch sind Halbmonde (Proliferation von Epithelzellen und Infiltration von Monozyten Monozyten Zellen des angeborenen Immunsystems/ Makrophagen Makrophagen Zellen des angeborenen Immunsystems in den Bowman-Raum) in den Glomeruli. Diese entstehen in der Regel durch eine immunologische Verletzung. Die Hauptmechanismen der immunologischen Verletzung werden in antiglomeruläre Basalmembran-(Anti-GBM)-RPGN, ANCA-assoziierte Vaskulitiden Vaskulitiden Vaskulitiden und Immunkomplex-vermittelte RPGN eingeteilt. Eine RPGN kann sich mit Hämaturie, Proteinurie und Ödemen, sowie Hypertonie Hypertonie Arterielle Hypertonie unterschiedlichen Ausmaßes manifestieren. Die Diagnostik erfolgt anhand der Klinik, sowie mittels Labortests, Bildgebung und Nierenbiopsie. Ein schneller Therapiebeginn ist essenziell für die Prognose der Erkrankung. Die Therapie richtet sich nach den verschiedenen Formen der RPGN und kann unter anderem aus Kortikosteroiden, Cyclophosphamid und Plasmapherese bestehen.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Die Rapid progressive Glomerulonephritis Glomerulonephritis Membranoproliferative Glomerulonephritis (RPGN) ist eine seltene Form der Glomerulonephritis Glomerulonephritis Membranoproliferative Glomerulonephritis und geht einher mit einer schweren glomerulären Erkrankung und fortschreitendem Verlust der Nierenfunktion innerhalb von Wochen bis Monaten.
Die meisten Fälle sind auf eine immunologische Verletzung der Glomeruli zurückzuführen.

Anti-glomeruläre Basalmembran (Anti-GBM)-Antikörperkrankheit: Halbmonde im Lichtmikroskop
Bild : „Crescents on light microscopy“ von Mavani GP, Pommier M., Win S., Michelis MF, Rosenstock J. Lizenz: CC BY 4.0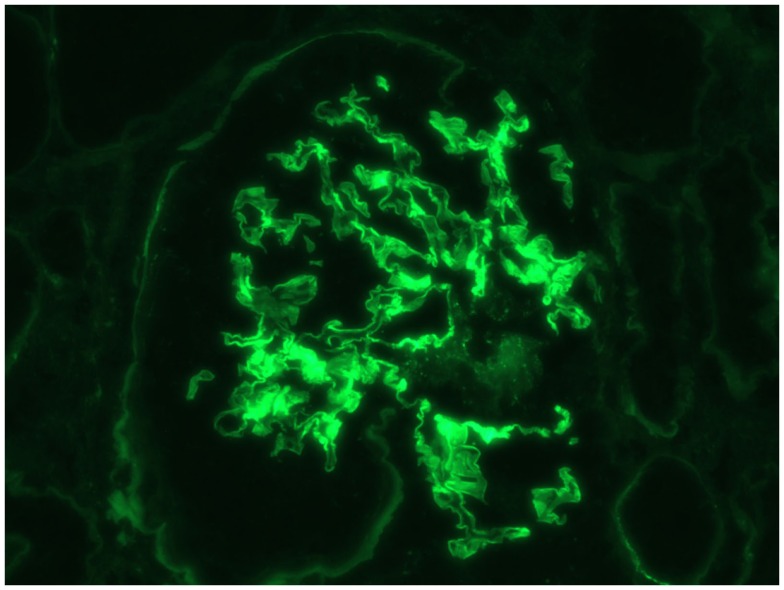
Anti-glomeruläre Basalmembran (Anti-GBM)-Antikörperkrankheit: IF-lineare Färbung von IgG
Bild: „Immunofluorescence Linear Staining of IgG“ von Mavani GP, Pommier M., Win S., Michelis MF, Rosenstock J. Lizenz: CC BY 4.0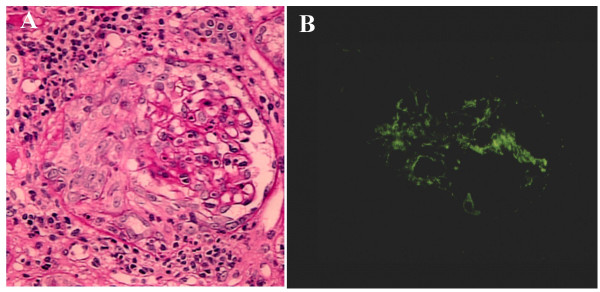
Infektiöse Endokarditis-induzierte (RPGN mit PAS-Färbung und IF:
A: PAS, das die zirkuläre und zelluläre Sichelbildung mit interstitiellen Nephritis zeigt
B: IF zeigt eine C3-positive Färbung im Mesangialbereich

Immunkomplex-vermittelte, halbmondförmige Glomerulonephritis mit Fibrin in den zellulären Halbmonden
Bild : „Cellular crescent“ von Syed R., Rehman A., Valecha G., El-Sayegh S. Lizenz: CC BY 3.0Ein schneller Beginn einer immunsuppressiven Therapie ist Prognose-entscheidend.


















