Ein epileptischer Anfall ist eine abnormale elektrische Aktivität der Neuronen Neuronen Nervensystem: Histologie in der Großhirnrinde, die sich je nach betroffener Gehirnregion auf verschiedene Weise manifestieren kann. Anfälle bestehen aus einem plötzlichen Ungleichgewicht, das zwischen den erregenden und hemmenden Signalen in kortikalen Neuronen Neuronen Nervensystem: Histologie auftritt und eine Nettoerregung erzeugt. Epiletische Anfälle und Epilepsien sind eine der häufigsten chronisch-neurologischen Erkrankungen im Erwachsenenalter. Die 2 Hauptklassen von Anfällen sind fokal oder generalisiert. Die Diagnose wird klinisch gestellt und basiert auf der Anamnese und körperlicher Untersuchung, kann aber durch EEG und andere Messinstrumente je nach Verdachtsdiagnose ergänzt werden. Wichtig ist die Beurteilung von Verdachtsdiagnosen eines epileptischen Anfalls, wie Synkope Synkope Synkope oder transistorische ischämische Attacke. Die Behandlung umfasst sowohl anfallsunterbrechende als auch präventive Medikamente. In selbstlimitierenden Fällen ohne Hinweis auf eine strukturelle Ätiologie sind möglicherweise keine medikamentösen Therapien erforderlich.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Epileptische Anfälle sind Episoden einer neurologischen Dysfunktion, die durch abnormale exzitatorische Aktivitäten von Neuronen Neuronen Nervensystem: Histologie verursacht werden, die durch eine plötzliche Änderung der Sinne, Wahrnehmung, motorischen Aktivität oder des Verhaltens gekennzeichnet sind.
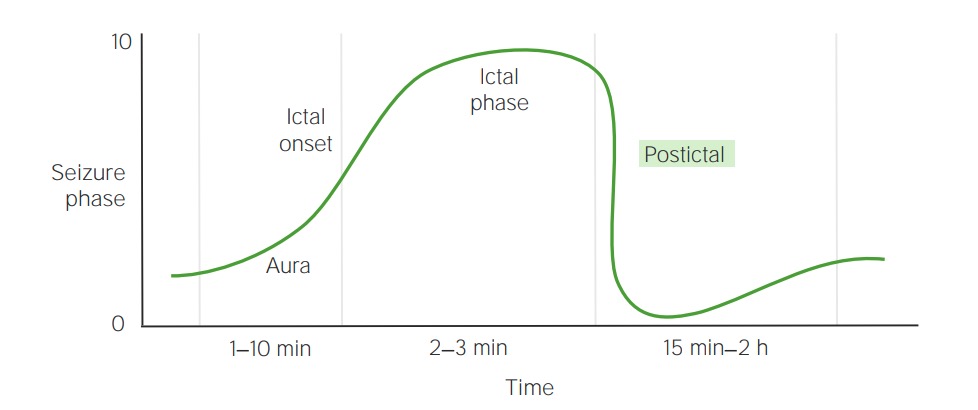
Zeitlicher Ablauf der Anfallsphasen
Bild von Lecturio.
Stufe 1: Kurzschluss im neuronalen Schaltkreis, der den 1. Schritt der Pathophysiologie von Krampfanfällen beschreibt.
AMPA: α-Amino-3-hydroxy-5-methyl-4-isoxazolpropionsäure
NMDA: N-Methyl-D-Aspartat

Stufe 2 der Pathophysiologie von Anfällen, Aktivierung der normalen Nachbarneurone:
Das Diagramm zeigt ein Membranpotential und Schritte, die es ermöglichen, dass überschüssiges K+ umgebende Zellen depolarisiert.

Stufe 3: Versagen der Inhibition in der Pathophysiologie von epileptischen Anfällen
Bild von Lecturio.| Ohne Einschränkung des Bewusstseins | Mit Einschränkung des Bewusstseins | Fokal zu bilateral tonisch-klonisch |
|---|---|---|
|
|
|
| Absence | Kurze generalisierte Starrperiode mit Bewusstseinseinschränkung, oft verwechselt mit Tagträumen, mit 3-Hz-Spike-and-Wave-EEG |
|---|---|
| Myoklonisch | Typischerweise fokale myoklonische Zuckungen mit kurzer Bewusstlosigkeit, aber ohne Konvulsionen und geringer postiktaler Beeinträchtigung |
| Tonisch | Fokale, isolierte Steifigkeit |
| Generalisiert tonisch-klonisch | Am häufigsten konvulsiv (aber auch nicht-konvulsiv möglich) mit Generalisierung und ohne Aura |
| Atonisch | „Drop-Attacken“ am häufigsten bei Kindern |

Krampfanfallsarten und Klassifikationen
Bild von Lecturio.Die initiale Bewertung und Stabilisierung konzentriert sich auf das Screening von Hypoglykämie Hypoglykämie Hypoglykämie, Hypoxie und hämodynamischer Instabilität.

Arten normaler elektrischer Aktivität auf einem Elektroenzephalogramm
Bild von Lecturio.
Elektrische Aktivität des Gehirns in einem EEG während eines Anfalls
Bild von Lecturio.| Absence | Krampfanfall | Synkope Synkope Synkope | PNES | |
|---|---|---|---|---|
| Labor |
|
|
|
|
| Bildgebung |
|
|
|
Keine erforderlich |
| Weitere Untersuchungen |
|
|
|
|
| Krampfanfall | Synkope Synkope Synkope | Transitorische ischämische Attacke | |
|---|---|---|---|
| Aura | Ja oder keine | Keine, Übelkeit und Erbrechen Erbrechen Erbrechen im Kindesalter, Schwindel | Keine |
| Haltung | Keine | Aufrecht | Keine |
| Beginn | Akut | Akut/variabel | Akut |
| Dauer | 1-2 Minuten | Sekunden bis Minuten | Minuten bis Stunden |
| Motorik | Variabel/tonisch-klonisch | Myoklonus Myoklonus Neurologische Untersuchung mit verlorenem Tonus | Defizite |
| Inkontinenz | Variabel | Sehr selten | Keine |
| EEG | Epileptiform | Generalisierte Verlangsamung | Fokale Verlangsamung oder generalisierte Verlangsamung mit posterioren Zirkulationsereignissen |
| Trauma | Variabel | Variabel | Normalerweise nicht |
| Post-Ereignis | Langsam/verwirrt | Schnell/alarmiert | Alarmiert |

Wirkmechanismen von Antiepileptika
VGCC: spannungsgesteuerte Calciumkanäle




















