Chorea Huntington ist eine fortschreitende neurodegenerative Erkrankung mit einem autosomal-dominanten Erbgang und einer schlechten Prognose. Sie wird durch Cytosin-Adenin-Guanin (CAG)-Trinukleotid-Wiederholungen im Huntingtin-Gen (HTT) verursacht. Das häufigste klinische Bild im Erwachsenenalter ist eine Bewegungsstörung, die als Chorea bekannt ist: abrupte, unwillkürliche Bewegungen des Gesichts, des Rumpfes und der Extremitäten. Psychiatrische und kognitive Symptome sind ebenfalls charakteristisch, im Verlauf der Erkrankung besteht ein erhöhtes Risiko für Depressionen und Suizid. Die Diagnose ist in erster Linie klinisch, oft kombiniert mit einer positiven Familienanamnese Familienanamnese Vorsorgeuntersuchungen und Prävention im Erwachsenenalter. Zur weiteren Bestätigung können genetische Tests durchgeführt werden. Die Betreuung der Patient*innen durch ein interdisziplinäres Team ist entscheidend für das Ziel, die Lebensqualität so lang wie möglich zu erhalten. Die Behandlung von Depression, Agitiertheit und Psychose hat für Patient*innen mit Morbus Huntington Vorrang vor der Behandlung von Chorea.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Chorea Huntington ist eine erbliche und fortschreitende neurodegenerative Erkrankung, die eine choreatische Bewegungsstörung, einen unsicheren Gang und geistigem Verfall verursacht.
Die Huntington-Krankheit wird durch eine autosomal-dominante genetische Veränderung des Huntingtin-Gens (HTT) auf dem Chromosom Chromosom Grundbegriffe der Genetik 4 verursacht.

Erhöhte Cytosin-Adenin-Guanin (CAG)-Triplett Wiederholung im Huntingtin-Gen (HTT) →Huntington-Krankheit
Bild : „Huntington`s disease patient“ von National Institute of Standards and Technology. Lizenz: Public Domain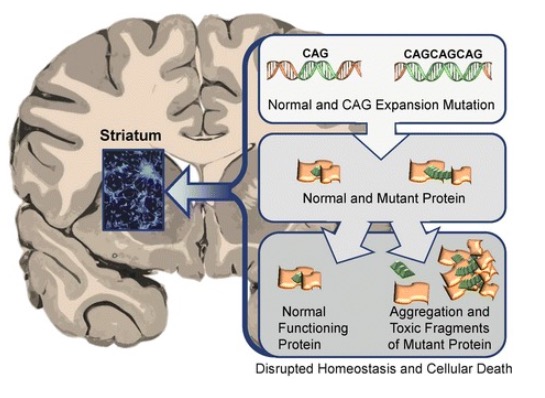
Abnorme toxische Wirkung von HTT-Proteinen auf das Striatum im Rahmen der Huntington-Krankheit, die zu Neurodegeneration und schließlich zum Tod der betroffenen Neuronen führt
Bild : „Neuronal changes in Huntington disease“ von Jaenisch R, Zhang F, Gage F. Lizenz: CC BY 4.0Die Huntington-Krankheit betrifft Generationen von Familienmitgliedern und erstreckt sich typischerweise über mehrere Jahrzehnte. Die Symptome können motorisch, kognitiv und verhaltensbedingt sein.
Die Therapie der Huntington-Krankheit umfasst medizinische und psychologische Unterstützung, um die Symptome zu behandeln und die Lebensqualität zu erhalten. Die Behandlung von Depression, Agitiertheit und Psychose hat für die Patient*innen Vorrang vor der Behandlung der Chorea.




















