Kardiomyopathien bezeichnen eine Gruppe von Herzmuskelerkrankungen, die mit strukturellen Veränderungen der Herzmuskulatur (Myokard) und einer beeinträchtigten systolischen und/oder diastolischen Funktion in Abwesenheit anderer Herzerkrankungen (koronare Herzkrankheit, Bluthochdruck, Herzklappenerkrankungen und angeborene Herzfehler) einhergehen. Die Liste der Ursachen ist umfangreich und reicht von familiären Störungen über Grunderkrankungen bis hin zu Infektionen. Betroffene stellen sich häufig mit Brustschmerzen Brustschmerzen Brustschmerzen, Atemnot Atemnot Dyspnoe (Atemnot/Luftnot), Herzklopfen und/oder Synkope Synkope Synkope vor. Einige Patient*innen können völlig asymptomatisch sein, während bei anderen ein plötzlicher Herztod Plötzlicher Herztod Plötzlicher Herzstillstand als erstes Anzeichen der Grunderkrankung auftreten kann. Die Diagnose wird durch EKG EKG Normales Elektrokardiogramm (EKG) und kardiale Bildgebung wie Echokardiographie und kardiale MRT MRT Magnetresonanztomographie (MRT) gestellt. Die Therapie umfasst Medikamente, die typischerweise zur Behandlung von Herzinsuffizienz verwendet werden, sowie die Behandlung möglicher Arrhythmien. In schweren Fällen kann eine Herztransplantation Herztransplantation Organtransplantation erforderlich sein.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Kardiomyopathien sind Erkrankungen, die die Struktur und Funktion des Herzmuskels (Myokard) ohne sekundäre Ursachen (z. B. koronare Herzkrankheit, Bluthochdruck, Herzklappenerkrankungen und angeborene Herzfehler) beeinträchtigen.
Ursachen variieren:
Ungefähr ⅓ der Fälle haben eine genetische Ursache.
Nach Morphologie:
Nach Ätiologie:
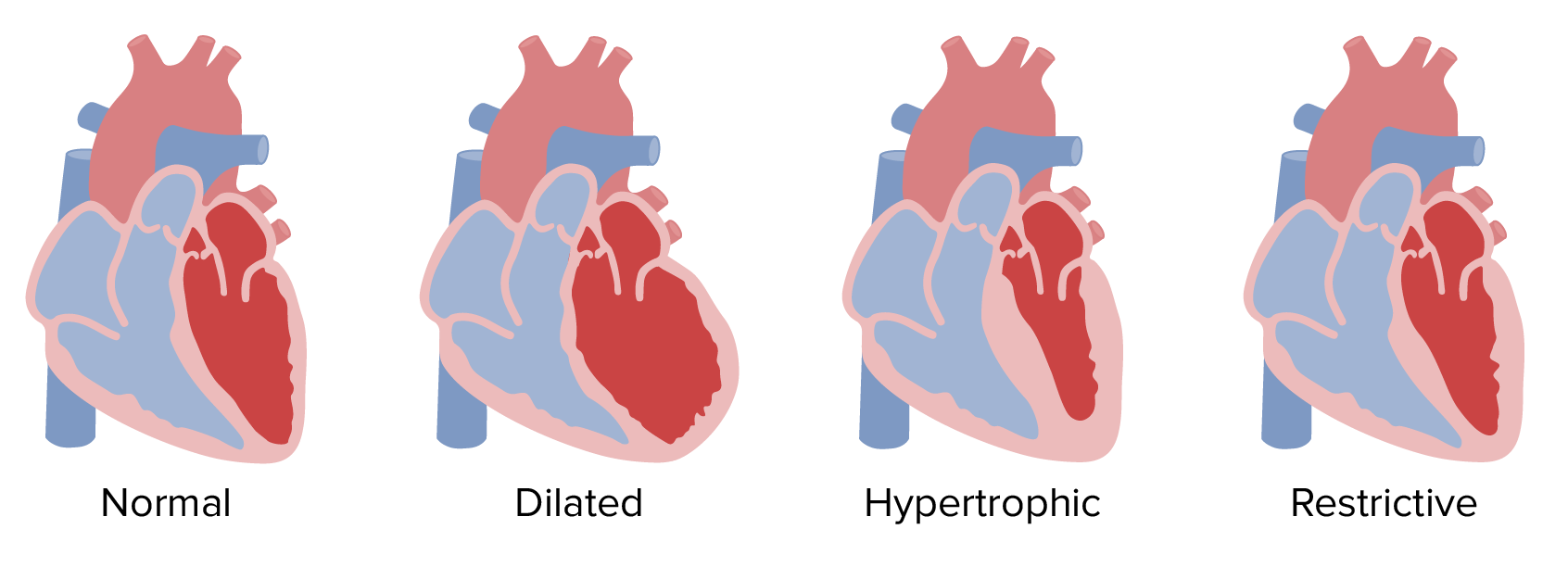
Morphologische Klassifikation der Kardiomyopathien
Bild von LecturioDas Myokard ist die dickste Schicht des Herzens.
Die Auswirkungen von Kardiomyopathien sind Strukturveränderung des Myokards:
Die strukturelle Veränderung des Herzens bewirkt je nach Krankheitsprozess eine systolische oder diastolische Dysfunktion:
Der Netto-Effekt ist eine insuffiziente Pumpleistung:
Der diagnostische Ansatz variiert je nach Art der Kardiomyopathie. Bei Herzinsuffizienz sollte stets eine Kardiomyopathie bedacht werden.
Therapieziele:
In therapierefraktären Fällen kann eine Herztransplantation Herztransplantation Organtransplantation angezeigt sein.
Komplikationen:
| Dilatative | Restriktive | Hypertrophe | Arrhythmogene rechtsventrikuläre | |
|---|---|---|---|---|
| Ätiologie |
|
|
|
|
| Klinik |
|
|
|
|
| Ergebnisse der körperlichen Untersuchung |
|
|
|
|
| Ejektionsfraktion Ejektionsfraktion Herzzyklus | ↓ | ↓ oder normal | ↑ oder normal | ↓ |
| L ventrikuläre diastolische Ausdehnung | ↑ | ↑ | ↓ | Normal |
| Linksventrikuläre Wandstärke | ↓ | Normal oder | ↑↑ | Normal |
| Vorhofgröße | ↑ | ↑ | ↑ | Normal |
| Diagnostik |
|
|
|
|
Ein vergrößertes Herz (Kardiomegalie) ist meist ein Symptom für eine andere Grunderkrankung und nicht eine eigenständige Krankheit. Es kann auf verschiedene Herz-Kreislauf-Probleme hinweisen, darunter Herzinsuffizienz, Herzklappenerkrankungen oder Kardiomyopathien. In manchen Fällen kann es zu lebensbedrohlichen Zuständen wie Herzrhythmusstörungen oder plötzlichem Herzversagen führen. Ein vergrößertes Herz ist daher ein Grund für weitere medizinische Untersuchungen, um die zugrundeliegende Ursache zu identifizieren und entsprechend zu behandeln.
In einigen Fällen kann sich ein vergrößertes Herz zurückbilden oder verbessern, vor allem wenn die zugrundeliegende Ursache effektiv behandelt wird. Zum Beispiel kann bei erfolgreicher Kontrolle des Bluthochdrucks oder bei Behandlung einer Herzinsuffizienz eine Verbesserung der Herzgröße und -funktion beobachtet werden. Die Möglichkeit der Rückbildung hängt jedoch von verschiedenen Faktoren ab, unter anderem dem Schweregrad der Erkrankung, der allgemeinen Gesundheit des Betroffenen und einem frühzeitigen Behandlungsbeginn.
Der Begriff „grenzwertig vergrößertes Herz“ weist auf eine leichte Zunahme der Herzgröße hin, die zwar nicht im Normalbereich liegt, aber auch nicht deutlich genug vergrößert ist, um als deutliche Kardiomegalie eingestuft zu werden. In solchen Fällen ist es besonders wichtig, die zugrundeliegende Ursache zu ermitteln und weitere diagnostische Tests durchzuführen, da eine grenzwertige Vergrößerung ein Frühzeichen für eine Erkrankung sein könnte, die behandelt werden sollte.




















