Hämolytische Anämien sind eine große Gruppe von Anämien, denen eine vorzeitige Zerstörung bzw. Hämolyse der zirkulierenden roten Blutkörperchen zugrunde liegt. Die Hämolyse kann innerhalb (intravasale Hämolyse) oder außerhalb der Blutgefäße (extravasale Hämolyse) auftreten. An der extravasalen Zerstörung von Erythrozyten Erythrozyten Erythrozyten sind Makrophagen Makrophagen Zellen des angeborenen Immunsystems der Leber Leber Leber, der Milz Milz Milz, des Knochenmarks und der Lymphknoten Lymphknoten Lymphsystem beteiligt. Zusätzlich können hämolytische Anämien nach der Art der Erythrozytenschädigung klassifiziert werden. Bei korpuskulären hämolytischen Anämien sind die Erythrozyten Erythrozyten Erythrozyten aufgrund einer vererbten Mutation strukturell verändert ( Hämoglobinopathien Hämoglobinopathien Anämie: Überblick und Formen) oder weisen einen Membrandefekt oder einen Stoffwechseldefekt auf. Ist nicht die strukturelle Veränderung, sondern ein anderer Faktor, wie Autoantikörper, ein mechanisches Trauma oder ein Krankheitserreger Ursache für die frühzeitige Zerstörung, wird die Anämie Anämie Anämie: Überblick und Formen als extrakorpuskuläre Hämolyse bezeichnet. Dabei handelt es sich fast immer eine erworbene Störung.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Intravasale Hämolyse
Extravasale Hämolyse
Die hämolytische Anämie Anämie Anämie: Überblick und Formen ist eine normozytäre Anämie Anämie Anämie: Überblick und Formen.

Normaler Lebenszyklus eines Erythrozyten
Bild von Lecturio.
Ikterus bei einem Kind mit G6PD-Mangel
Bild: „Jaundice“ von Sab3el3eish. Lizenz: CC BY 3.0Allgemein
Intravasale Hämolyse
Extravasale Hämolyse
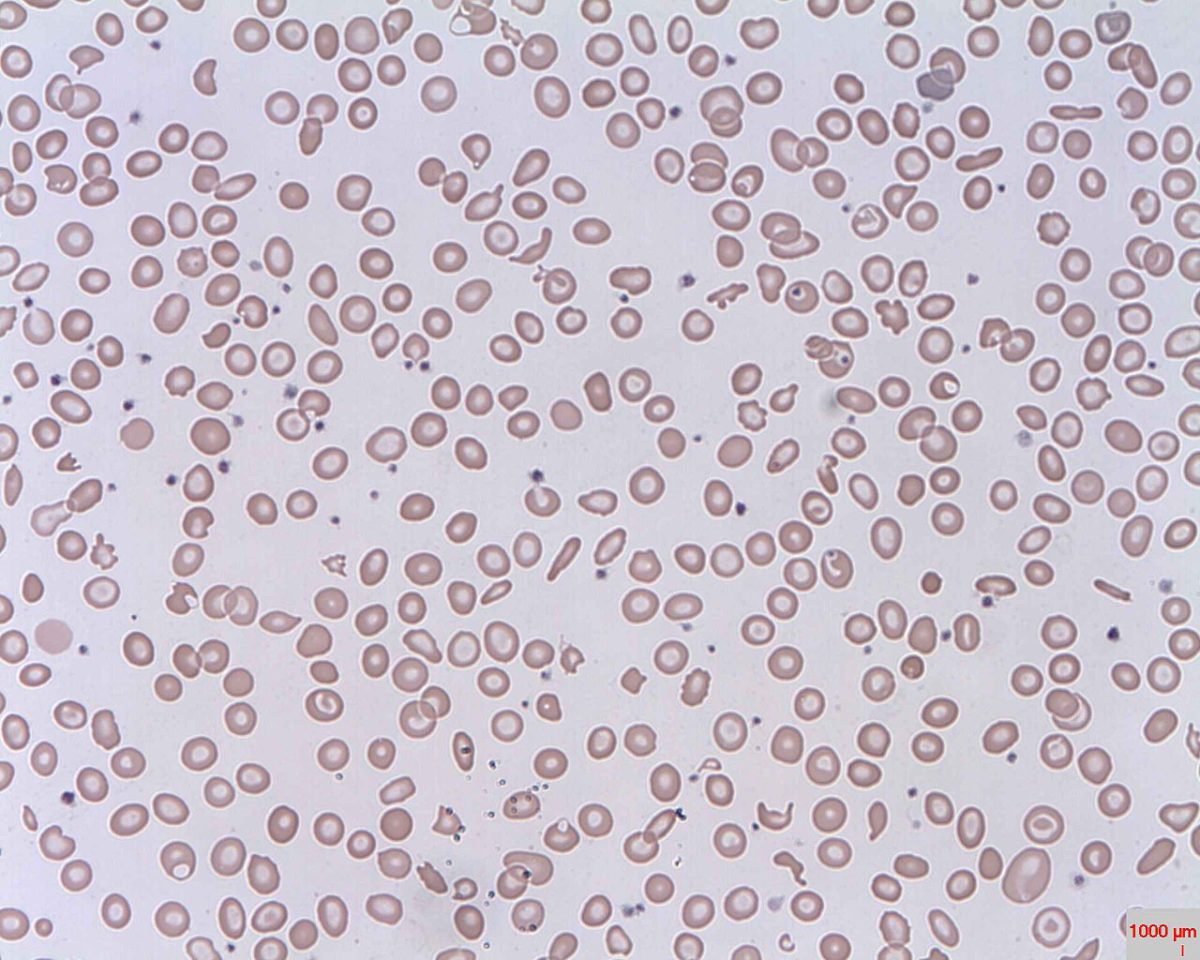
Fragmentozyten (fragmentierte Erythrozyten) bei intravasaler Hämolyse
Bild : „Schistocytes“ von Prof. Osaro Erhabor. Lizenz: Public Domain
Hämosiderinurie bei intravasaler Hämolyse
Bild : „Hemosiderinuria. Perls’ reaction weakly positive in the urine.“ von Salido, Eduardo & Cabañas, Valentín & Berenguer, Mercedes & Macizo, María & Candel, Faustino & Pérez-López, Raúl & Moraleda, Jose. (2014). Serologische Befunde bei einem Kind mit paroxysmaler Kältehämoglobinurie. Fallberichte in der Medizin. 2014, bearbeitet von Lecturio.
Patient*in mit CLL stellt sich mit AIHA mit Wärmeantikörpern vor. Blutausstrich zeigt Sphärozyten.
Bild : „CLL with Autoimmune Hemolytic Anemia“ von Ed Uthman aus Houston, TX, USA. Lizenz: CC BY 2.0



















