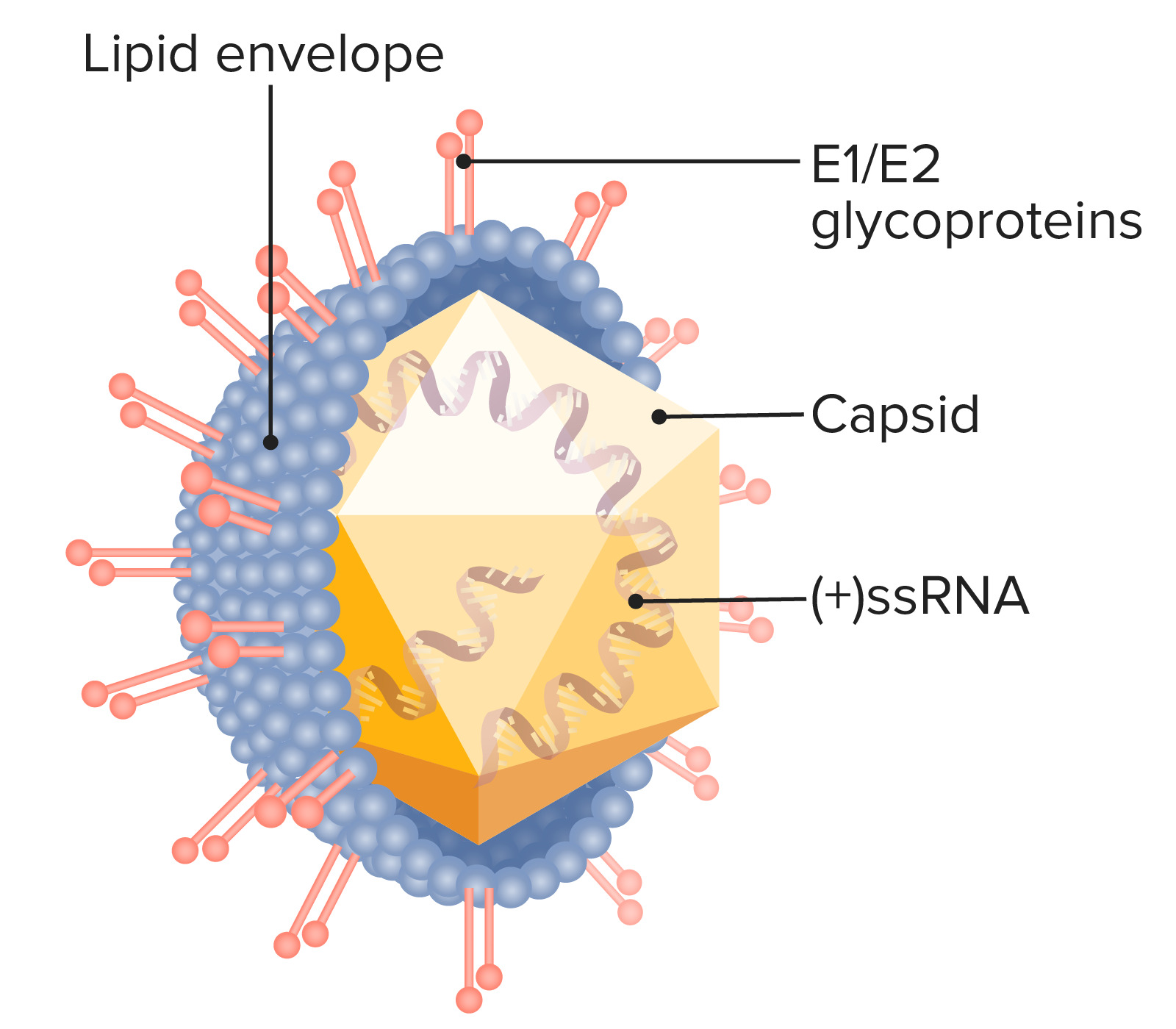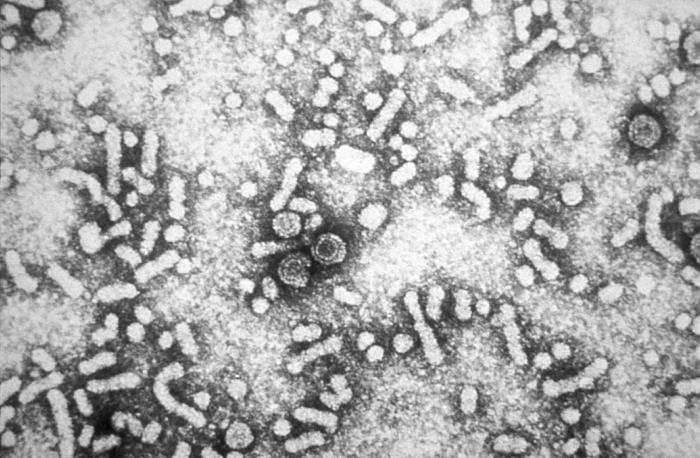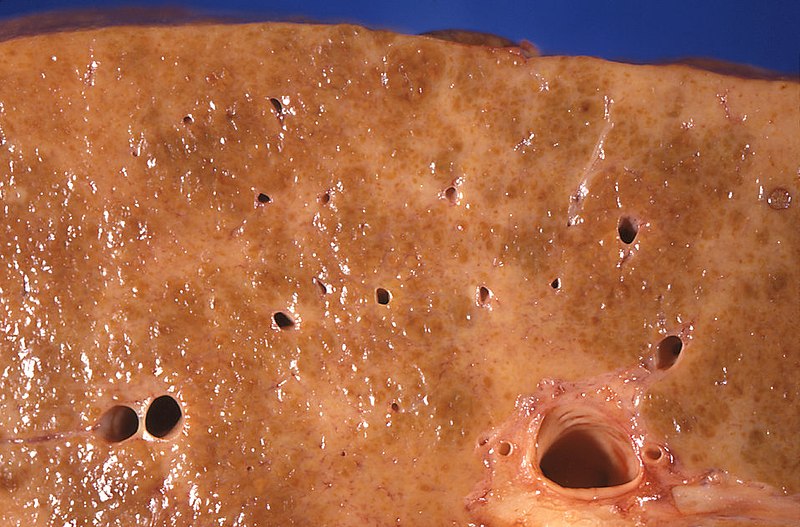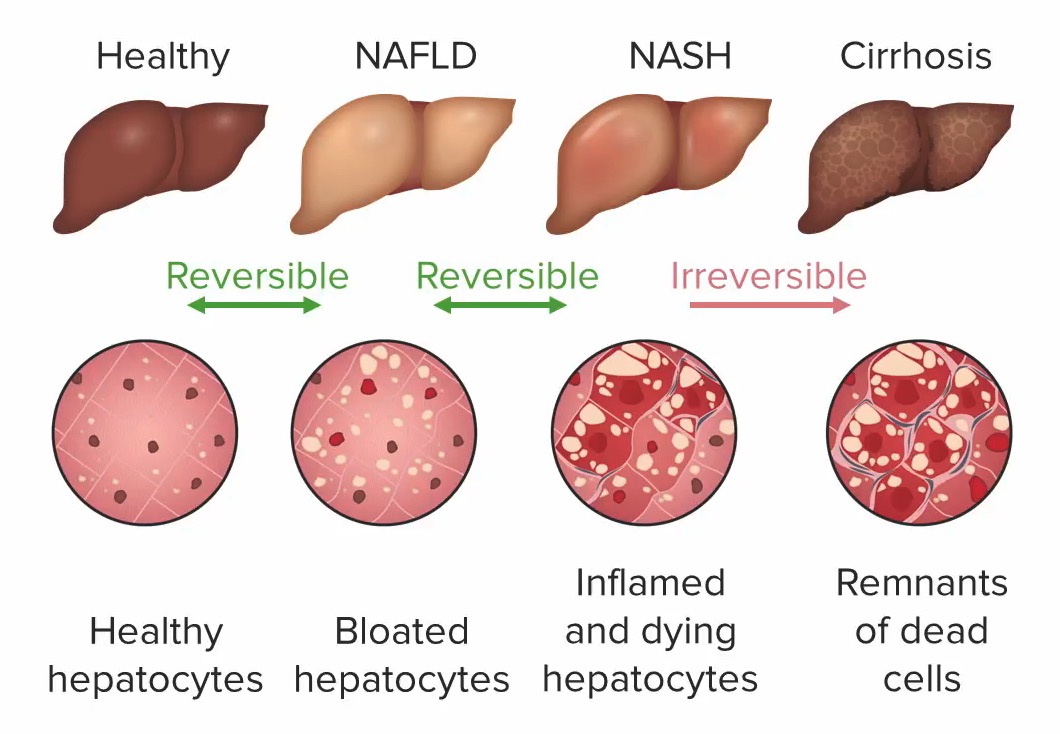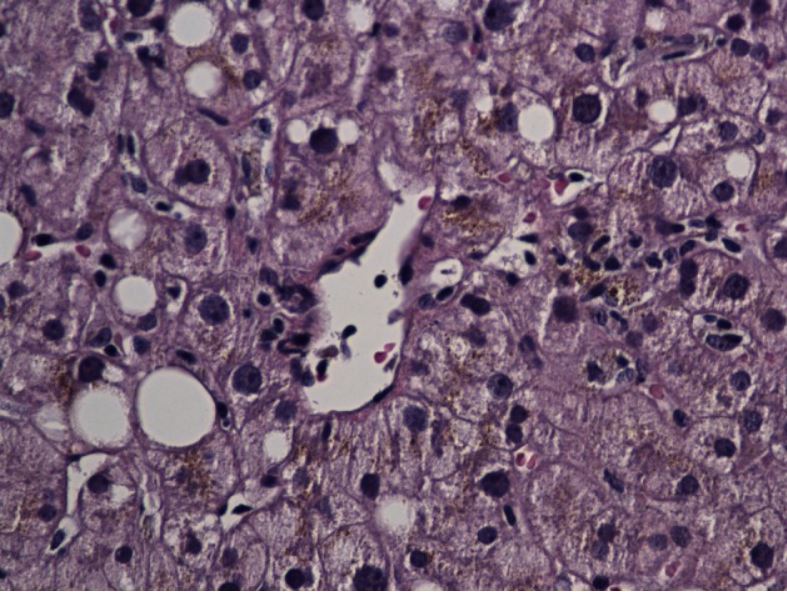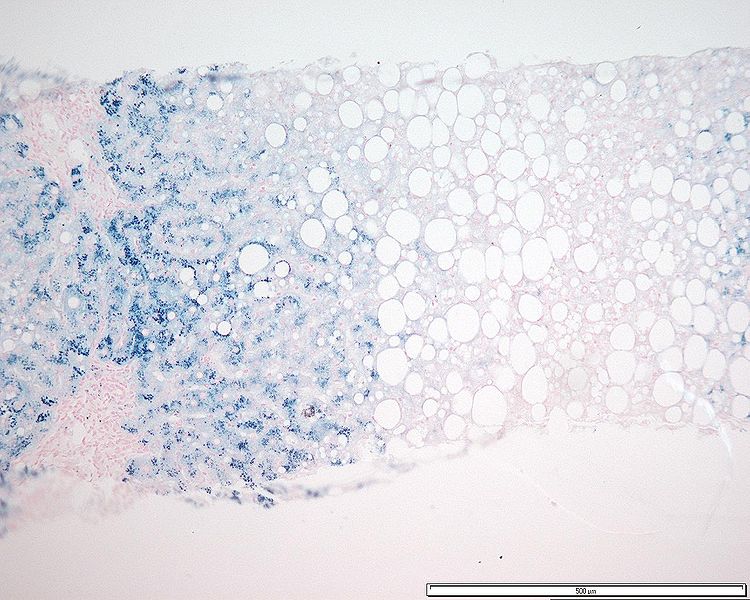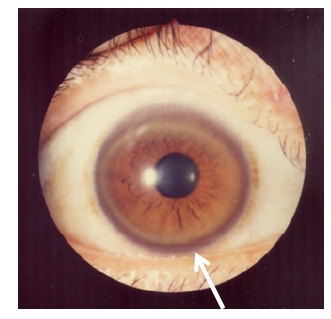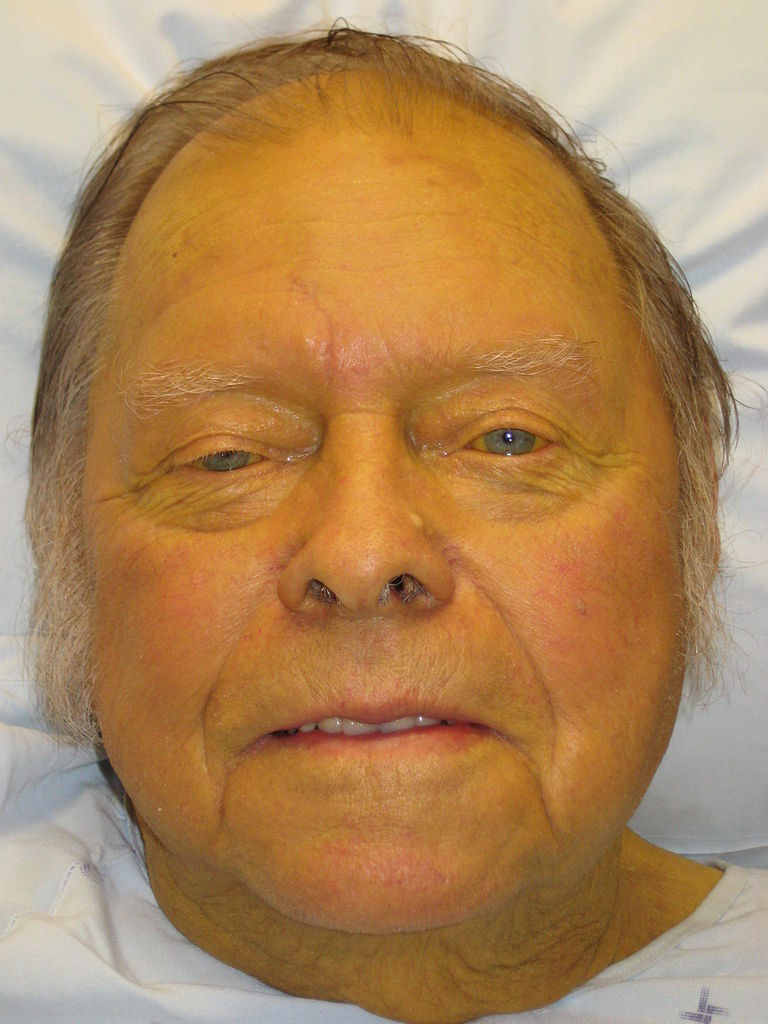
Disorders of the Hepatobiliary System
by Richard Mitchell, MD, PhDThis course provides a comprehensive study of hepatobiliary system disorders and their management. Understanding hepatobiliary disorders is crucial for medical professionals as these conditions are prevalent and can have profound impacts on a patient's quality of life.
Prior knowledge of human anatomy and physiology is recommended to fully comprehend the course materials. This course bridges the foundational understanding of hepatobiliary system function with clinical applications, enabling learners to diagnose and manage these disorders effectively in a healthcare setting.
Prior knowledge of human anatomy and physiology is recommended to fully comprehend the course materials. This course bridges the foundational understanding of hepatobiliary system function with clinical applications, enabling learners to diagnose and manage these disorders effectively in a healthcare setting.
Course Details
- Videos 21
- Duration 2:41 h
- Quiz questions 82
- Concept Pages 17